Microdiversity of Phenol Hydroxylase Genes Among Phenol-Degr
| 论文类型 | 技术与工程 | 发表日期 | 2008-01-01 |
| 来源 | 第三届环境模拟与污染控制学术研讨会 | ||
| 作者 | XUELI,ZHANG,PINGPING,GAO,QUNFA | ||
| 关键词 | microdiversity, phenol hydroxylase, horizontal gene transfer, activated sludge | ||
| 摘要 | Enterobacterial repetitive intergenic consensus (ERIC)-PCR fingerprinting classified 97 phenol-degrading isolates with identical amplified ribosomal DNA restriction analysis (ARDRA) patterns into 6 genotypic groups. The 16S rRNA gene of the representative | ||
XUELI ZHANG1, PINGPING GAO1, QUNFANG CHAO1, LINGHUA WANG1, ERIC SENIOR2, LIPING ZHAO1*
1Laboratory of Molecular Microbial Ecology and Ecogenomics, College of Life Science and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China; 2Centre for Advanced Water Technology, Singapore Utilities International Pte Ltd, Innovation Centre (NTU), Singapore 637723
*Address of the corresponding author: Liping Zhao Email: lpzhao@sjtu.edu.cn
Abstract
Enterobacterial repetitive intergenic consensus (ERIC)-PCR fingerprinting classified 97 phenol-degrading isolates with identical amplified ribosomal DNA restriction analysis (ARDRA) patterns into 6 genotypic groups. The 16S rRNA gene of the representative isolate of each group had higher than 99.47% common identity with each other and higher than 98% identity with the type strain of Alcaligenes faecalis. PCR-TGGE (temperature gradient gel electrophoresis) analysis of the genes of the largest subunit of the multi-component phenol hydroxylase (LmPH) in each isolate followed with sequencing showed that isolates within each ERIC-PCR group had identical LmPH gene sequences. Among the six different ERIC-PCR groups, two were found to harbor two different LmPH genes encoding low- and high-Ks (affinity constants) phenol hydroxylases, and the low-Ks type LmPH was identical in sequence with one predominant LmPH of the parental activated sludge. Three ERIC-PCR groups had only the high-Ks type and one had no sequence similar to the known LmPHs. Our work suggests that there is no correlation between the phylogenetic groupings of phenol-degrading bacteria and their LmPH genotypes possibly due to extensive horizontal gene transfer of this functional gene.
Keywords: microdiversity, phenol hydroxylase, horizontal gene transfer, activated sludge
Introduction
A wide diversity of phenol-degrading bacteria, including Pseudomonas (Futamata et al., 2001a; Watanabe et al., 1996), Ralstonia (Futamata et al., 2001a; Watanabe et al., 1996), Acinetobacter (Ehrt et al, 1995), Comamonas (Watanabe et al., 1996), Burkholderia (Watanabe et al., 1996) and Variovorax (Futamata et al., 2001a), have been isolated from natural or engineered environments, and their functional and genetic diversity have been characterized extensively (Futamata et al., 2001a; Futamata et al., 2001b; Watanabe et al., 1996; Watanabe et al., 1998).
Phenol hydroxylase is responsible for converting phenol to catechol, which is the initial and rate-limiting step in phenol degradation pathways (Hino et al., 1998). Both single component and multi-component types of this enzyme have been identified with the latter recognized as predominant in natural environments (Futamata et al., 2001a; Peters et al., 1997; Watanabe et al., 1998). The DNA fragment encoding the largest subunit of the multi-component phenol hydroxylase (LmPH) has been used as a molecular marker to assess the functional and genetic diversities of phenol-degrading bacteria in the environment (Watanabe et al., 1998; Watanabe et al., 2002). Phylogenetic groupings of the phenol-degrading bacteria based on the amino acid sequences of the LmPH genes were correlated with the physiological groupings based on their whole cell kinetic traits of low- and high-Ks (affinity constants) for phenol degradation. Most of the strains so far discovered with the high-Ks LmPH gene are affiliated with the gamma subclass of the class Proteobacteria,especially the genus Pseudomonas, while all of the strains in the low-Ks cluster are from the beta subclass (Watanabe et al., 2002). However, the assessment of diversity has been carried out mostly at the species level or higher while microdiversity of phenol-degrading bacteria has rarely been addressed due to the limited number of isolates characterized for each species in this type of work.
In our previous work, we have obtained 97 phenol-degrading isolates with identical amplified ribosomal DNA restriction analysis (ARDRA) patterns by direct plating of activated sludge mixed liquor from an aeration tank of an industrial coking wastewater treatment plant onto agar plates containing raw feed water as medium (Gao et al., 2003). These phylogenetically closely related isolates constituted excellent material for assessing microdiversity of phenol hydroxylase genes of phenol degrading populations isolated from a single habitat.
In the present study, we used both enterobacterial repetitive intergenic consensus (ERIC)-PCR fingerprinting and LmPH gene to analyze these 97 phenol-degrading isolates. Both low- and high-Ks types of LmPH genes were found in these isolates, and even in a single isolate. Thus we suggest that there is no correlation between the phylogenetic groupings of phenol-degrading bacteria and their LmPH genotypes.
2. Materials and methods
2.1. Sample Collection and Bacterial Isolates
Samples of activated sludge mixed liquor were obtained previously from an aeration tank of an industrial coking wastewater treatment plant in Taiyuan, China (Gao et al., 2003). 97 phenol-degrading isolates with identical ARDRA patterns were obtained by direct plating of a serially diluted activated sludge sample onto a feeding water-based medium.
2.2. ERIC-PCR Fingerprinting
The ERIC-PCR fingerprint of each isolate was obtained by PCR amplification of its genomic DNA with primers ERIC1R and ERIC2. The agarose gel profiles were visualized with an UVI gel documentation system (UVItec, Cambridge). A dendrogram was constructed based on the Dice similarity coefficient with unweighted pair group method clustering (UPGMA) with UVI band/map software (UVItec).
2.3. Sequencing of 16S rRNA Gene
Universal primers P0 and P6 (Di Cello et al., 1997) were used to amplify the 16S rRNA gene of the representative isolate of each ERIC-PCR group. Amplified 16S rRNA gene fragments were cloned with pGEM-T easy vector (Promega) into Escherichia coli DH5a. Three white colonies of each isolate were sequenced.
2.4. LmPH Gene Amplification
Primers Phe149GC and Phe212 as described previously (Watanabe et al., 1998) were used for PCR amplification of the LmPH gene, with a product size of 249 bp (including the GC clamp), from the 97 phenol-degrading isolates and the parental activated sludge DNA.
2.5. TGGE Analysis of the LmPH Gene
Parallel TGGE was performed with a TGGE-Mini system (Biometra) as described by the manufacturer. LmPH gene amplification products (249 bp) were electrophoresed in gels containing 8% acrylamide/bis (37.5:1), 8 M urea, and 20% formamide with a TAE buffer system, at a constant voltage of 200 V for 3 hours, applying a thermal gradient of 40-60°C. Subsequently, the gel was stained with AgNO3 as described by the manufacturer.
2.6. Sequencing of TGGE Bands
Each gel slice that contained a DNA band was excised and transferred into a sterile 1.5 ml Eppendorf tube, which contained 50 ml of sterile TE buffer, for elution of DNA fragments at 4℃ for 16 h. 1 ml supernatant was subjected to a second PCR under the same conditions as above. The PCR products were purified and sequenced as described above.
2.7. Hybridization Analysis
Two LmPH gene fragments (L-46d and L18) were DIG labelled with a DIG DNA labeling and detection kit (Roche, Germany). They were then used as probes to hybridize the genomic DNA of IS-17 under low hybridization stringency.
2.8. LmPH Gene Diversity in Parent Activated Sludge Sample
LmPH genes were amplified directly from activated sludge DNA and purified, ligated and transformed as described above. Twenty-four colonies were picked and sequenced. Coverage of the clone library was calculated using Good’s formula.
2.9. Sequence Analysis
The 16S rRNA and LmPH gene sequences were analyzed by the BLAST program to search for the most closely related sequences. Corrected evolutionary distances were computed using the correction described by Kimura. Phylogenetic trees were constructed by the neighbor-joining method. Bootstrap values were calculated for 100 phylogenetic trees with the Clustal X software.
3. Results
3.1. ERIC-PCR based genomic fingerprinting
6 distinct ERIC-PCR patterns were recognized with the abundance of each group quite different (Table 1). The ERIC-PCR patterns were identical within each of the group, and the pattern of the representative isolate of each group was depicted in Fig. 1a. E3 was most significantly different from the others (Fig. 1b).
3.2. 16S rRNA Gene Sequence Analysis
The identities between each pair of the six isolates were 99.47% or higher, indicative of close phylogenetic relationships at the species level, and these increased to between 99.6 and 99.8% when IS-17 (E3) was omitted. All the six isolates were, tentatively, affiliated to Alcaligenes faecalis, with 98.03% to 98.16% 16S rRNA gene sequence identities to type strain of A. faecalis (ATCC 8750).
3.3. LmPH Gene (Shorter Fragment) Analysis
All the isolates having identical ERIC-PCR patterns also showed identical TGGE profiles (data not shown). The LmPH gene was not amplified from E3 group. Three ERIC-PCR groups (E2, E5, and E6) had one TGGE band, indicating a single LmPH gene. To our surprise, two ERIC-PCR groups (E1 and E4) recorded two TGGE bands (c and d in Fig. 2) indicative of two LmPH gene fragments with different sequences. Sequencing of the TGGE bands of 1-5 isolates within each ERIC-PCR group further confirmed that isolates within each ERIC-PCR group had identical LmPH gene sequences. The LmPH gene sequence of each ERIC-PCR group was analyzed by its representative isolate hereafter (Table 1).
L-46d (from position 287 to 495) was found to have an identical nucleotide sequence with L-33d. In contrast, there were one to five nucleotide mismatches among L-18 (from position 287 to 495), L-67, L-92, L-33c and L-46c, but their deduced amino acid sequences were identical.
Two predominant bands (a and b) were visible in the TGGE profile of the activated sludge sample (Lane 1 of Fig. 2). The two LmPH gene sequences (L-ASa, accession no. AY346149 and L-ASb, accession no. AY346150) corresponding to bands a and b shared 79% homology with each other and L-ASb was found to have identical nucleotide sequence with L-33d and L-46d. LmPH gene clone library of the activated sludge was also constructed with 24 clones randomly picked for sequencing. Eighteen clones had an identical nucleotide sequence with L-ASa while 5 clones had an identical nucleotide sequence with L-ASb. The coverage of this clone library was 96%. This confirmed that L-ASa and L-ASb were predominant in the activated sludge.
Two major types of LmPH genes were thus identified for the isolates and the activated sludge sample. The first type, including L-18, L-67, L-92, L-33c and L-46c, was most closely related to the phenol hydroxylase of Acinetobacter radioresistens (AF521658) with 94% identity. The second type consisted of L-33d, L-46d, L-ASa and L-ASb. Tbc1D monooxygenase of Burkholderia cepacia JS150 (AF282897) was most closely related with this type with 86% similarity with L-ASa and 92% similarity with the others.
3.4. Hybridization Analysis
Since the LmPH gene was not amplified from isolates in E3 type, we hybridized the genomic DNA of its representative isolate, IS-17, with the two amplified LmPH gene fragments, L-46d and L-18, under low hybridization stringency. No signals were detected with either probe, while the two amplified LmPH gene fragments could hybridize with each other (data not shown), which suggested that the phenol hydroxylase genes of isolates in E3 were significantly different from those of the other isolates.
4. Discussion
High resolution molecular fingerprinting methods, such as randomly amplified polymorphic DNA (RAPD), repetitive extragenic palindromic (REP)-PCR, and ERIC-PCR, have been used widely to study microdiversity (Di Cello et al., 1997; Schloter et al., 2000). However, they can only distinguish phylogenetically closely related bacteria, with very limited genetic and ecological information to provide. On the other hand, protein-coding genes were also suggested to provide a better opportunity for distinguishing very closely related ecological populations than 16S rRNA genes (Palys et al., 1997). Unfortunately, very few ecologically distinct populations that are indistinguishable by 16S rRNA have been surveyed for variation at protein-coding gene loci (Palys et al., 1997). In our work, both ERIC-PCR fingerprinting and a protein-coding functional gene (LmPH) were used to access the microdiversity among a group of phylogenetically closely related phenol-degrading isolates. The 97 isolates could be classified into 6 genotypic groups by ERIC-PCR fingerprinting, while they had only 3 different LmPH genotypes. All the isolates with identical ERIC-PCR patterns had identical LmPH genes. However, isolates with different ERIC-PCR patterns could have either identical or different LmPH genes. Our results suggest that ERIC-PCR fingerprinting had a higher resolution power than the LmPH gene when accessing the microdiversity of phenol-degrading bacteria, while the latter one may be more ecologically relevant.
Because only a limited number of phenol-degrading isolates of each single species were characterized in early works, microdiversity and its ecological significance remain to be elucidated. -Microdiversity seems to be a general phenomenon in the microbial world (Schloter et al., 2000; Fuhrman et al., 1998; Jaspers et al., 2001; Moore et al., 1998). Most of the previous microdiversity investigations used strains isolated from different ecosystems and it was found that microdiversity was related to many factors such as spatial separation, habitat differences and specific bacterium-host interactions (Schloter et al, 2000). In contrast, our studied samples came from a single ecosystem in which the environmental factors, particularly the phenol concentration of the feed water, fluctuated markedly throughout the year. It is not surprising, therefore, that a set of bacteria with different phenol degrading kinetics can co-exist within this habitat since it has been suggested that the coexistence and distribution of multiple ecotypes permits the survival of the population as a whole over a broader range of environmental conditions than would be possible for a homogeneous population (Moore et al., 1998). In our case, the wastewater composition is very complex having many other aromatic compounds together with phenol. Since phenol-degrading bacteria can also metabolize many of these pollutants, we suggest that there might be many ecological niches for the different ecotypes to coexist and maintain the functional stability of the population.
Watanabe et al. demonstrated that phylogenetic groupings of phenol-degrading bacteria were correlated with physiological groupings based on their LmPH gene amino acid sequences and whole cell kinetic traits (Watanabe et al., 2002). However, extensive analysis of the 97 phylogenetically closely related isolates in this work revealed the presence of isolates harboring the high-Ks LmPH gene and isolates with both the low- and high-Ks types. Thus, the relationship between phylogenetic groupings and their phenol hydroxylase gene types is questionable in light of this result since both LmPH genotypes can exist in a single species, even in a single bacterium.
It has not been reported that one bacterium possessed different LmPH genes. Ehrt et al proposed the presence of two differentially regulated catechol 1,2-dioxygenase genes which are cotranscribed with phenol hydroxylase genes in Acinetobacter calcoaceticus NCIB8250 (Ehrt et al., 1995). Our work is the first to demonstrate that one strain can harbor two different LmPH genes. Furthermore, the low-Ks type LmPH gene was found to be predominant in the parental activated sludge sample while the high-Ks type was rarely detected. This suggests that the low-Ks type LmPH gene (L-ASb) in the parental activated sludge was not from those isolates having two LmPH genes (E1 and E4), but from other predominant phenol-degrading bacteria. We can imagine a natural scenario in which isolates of A. faecalis in this activated sludge system had only high-Ks LmPH gene originally. Some of them acquired the low-Ks LmPH gene from other bacteria via horizontal gene transfer under the selection pressure to become more adapted to the highly fluctuating environment.
Conclusion
Our work is the first to investigate the microdiversity of phenol hydroxylase of phylogenetically closely related phenol-degrading populations isolated from a single habitat. Two different LmPH genes corresponding to two different phenol-degrading kinetics were found in these phylogenetically closely related isolates, and even in a single isolate. We suggest that there is no correlation between the phylogenetic groupings of phenol-degrading bacteria and their LmPH genotypes possibly due to extensive horizontal gene transfer of this functional gene.
References:
Di Cello F., Bevivino A., Chiarini L., Fani R., Paffetti D., Tabacchioni S. and Dalmastri C. (1997). Biodiversity of a Burkholderia cepacia population isolated from the maize rhizosphere at different plant growth stages. Appl. Environ. Microbiol., 63, 4485-4493.
Ehrt S., Schirmer F. and Hillen W. (1995). Genetic organization, nucleotide sequence and regulation of expression of genes encoding phenol hydroxylase and catechol 1,2-dioxygenase in Acinetobacter calcoaceticus NCIB8250. Mol. Microbiol., 18, 13-20.
Fuhrman J.A. and Campbell L. (1998). Microbial microdiversity. Nature, 393, 393-394.
Futamata H., Harayama S. and Watanabe K. (2001). Diversity in kinetics of trichloroethylene-degrading activities exhibited by phenol-degrading bacteria. Appl. Microbiol. Biotechnol., 55, 248-253.
Futamata H., Harayama S. and Watanabe K. (2001). Group-specific monitoring of phenol hydroxylase genes for a functional assessment of phenol-stimulated trichloroethylene bioremediation. Appl. Environ. Microbiol., 67, 4671-4677.
Gao P., Chen Y., Liu B., Zhang X. and Zhao L. (2003). Isolation of novel phenol-degrading bacteria from activated sludge using feed water medium (FWM). Chin. J. Appl. Environ. Biol., 9, 189-192.
Hino S., Watanabe K. and Takahashi N. (1998). Phenol hydroxylase cloned from Ralstonia eutropha strain E2 exhibits novel kinetic properties. Microbiology, 144 ( Pt 7), 1765-1772.
Jaspers E., Nauhaus K., Cypionka H. and Overmann J. (2001). Multitude and temporal variability of ecological niches as indicated by the diversity of cultivated bacterioplankton. FEMS Microbiol. Ecol., 36, 153-164.
Moore L.R., Rocap G. and Chisholm S.W. (1998). Physiology and molecular phylogeny of coexisting Prochlorococcus ecotypes. Nature, 393, 464-467.
Palys T., Nakamura L.K. and Cohan F.M. (1997). Discovery and classification of ecological diversity in the bacterial world: the role of DNA sequence data. Int. J. Syst. Bacteriol., 47, 1145-1156.
Peters M., Heinaru E., Talpsep E., Wand H., Stottmeister U., Heinaru A. and Nurk A. (1997). Acquisition of a deliberately introduced phenol degradation operon, pheBA, by different indigenous Pseudomonas species. Appl. Environ. Microbiol., 63, 4899-4906.
Schloter M., Lebuhn M., Heulin T. and Hartmann A. (2000). Ecology and evolution of bacterial microdiversity. FEMS Microbiol. Rev., 24, 647-660.
Watanabe K., Hino S., Onodera K., Kajie S. and Takahashi N. (1996). Diversity in kinetics of bacterial phenol-oxygenating activity. J. Ferment. Bioeng., 81, 560-563.
Watanabe K., Teramoto M., Futamata H. and Harayama S. (1998). Molecular detection, isolation, and physiological characterization of functionally dominant phenol-degrading bacteria in activated sludge. Appl. Environ. Microbiol., 64, 4396-4402.
Watanabe K., Futamata H. and Harayama S. (2002). Understanding the diversity in catabolic potential of microorganisms for the development of bioremediation strategies. Antonie van Leeuwenhoek, 81, 655-663.
Table1. Classification of the 97 phenol-degrading isolates based on both ERIC-PCR fingerprinting and LmPH gene.
| ERIC-PCR group | Number of isolates | Representative isolate | LmPH gene (accession number) | LmPH group |
| E1 | 57 | IS-46 | L-46c (AY346145) L-46d (AY346146) | III I |
| E2 | 12 | IS-18 | L-18 (AY346142) | III |
| E3 | 8 | IS-17 | / | / |
| E4 | 1 | IS-33 | L-33c (AY346143) L-33d (AY346144) | III I |
| E5 | 15 | IS-67 | L-67 (AY346147) | III |
| E6 | 4 | IS-92 | L-92 (AY346148) | III |

FIG. 1. ERIC-PCR banding patterns (a) and cluster analysis (b) of the phenol-degrading isolates. The ERIC-PCR patterns are negative images of an ethidium-bromide stained gel. The UPGMA dendrogram is based on the ERIC-PCR patterns of the 6 ERIC-PCR groups with the scale bar representing the Dice similarity coefficient of the ERIC-PCR patterns.
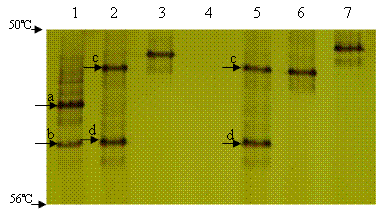
FIG. 2. TGGE profiles of the LmPH gene fragments of the parental activated sludge sample and the representative isolate of each ERIC-PCR group. Lane 1, activated sludge sample; Lanes 2 to 7, isolates IS-46 (E1), IS-18 (E2), IS-17 (E3), IS-33 (E4), IS-67 (E5) and IS-92 (E6), respectively.
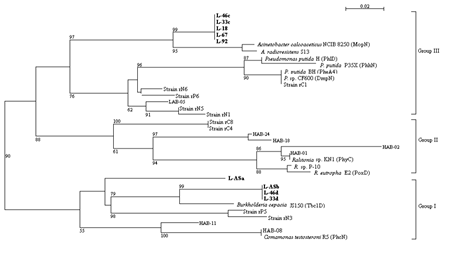
FIG. 3. Unrooted neighbor-joining tree based on the partial amino acid sequences of the LmPHs identified in this paper (bold face) and previously reported representative LmPHs retrieved from GenBank. The numbers at the branch nodes are bootstrap values (per 100 trials) with only values greater than 50 indicated. The bar represents 0.02 substitution per amino acid site.
相关推荐
- 枣椰仁颗粒活性碳及其在多介质过滤器中余氯穿透曲线动态建模(英文)
- Micro-Polluted Surface Water Treatment by PAC-MBR Process
- Study on adsorption and biochemical regeneration of clinoptilolite for ammonia removal
- A_O工艺与分段进水两种生物脱氮工艺的比较研究_英文
- Biodegradation of 2,4-dichlorophenol in an air-lift honeycomb-like ceramic reactor
- Bioadsorption of pentachlorophenol (PCP) from aqueous solution by activated sludge biomass
论文搜索
月热点论文
论文投稿
很多时候您的文章总是无缘变成铅字。研究做到关键时,试验有了起色时,是不是想和同行探讨一下,工作中有了心得,您是不是很想与人分享,那么不要只是默默工作了,写下来吧!投稿时,请以附件形式发至 paper@h2o-china.com ,请注明论文投稿。一旦采用,我们会为您增加100枚金币。



